当前,全球清洁能源转型的步伐日益加快,世界各国对氢能发展寄予厚望,而电解水制氢有望成为清洁氢气的主要生产方式。目前,大部分已报道的电解水体系均基于资源有限的纯水,忽略了占地球水总量的97%的海水资源。由于海水成分复杂,尤其是氯离子浓度较高,电解海水制氢催化剂的构建面临诸多挑战。因此,亟需开发耐盐腐蚀且兼具高活性、高稳定性的电解海水阴阳极催化剂。
近日,青岛科技大学生态化工国家重点实验室培育基地王磊教授团队在电解海水催化剂研究方面取得重大进展,相关成果先后发表在advanced energy materials (2024, 2400975;if:24.4),advanced functional materials (adfm.202412522;if:18.5),angewandtechemieinternationaledition(2024, e202316319;if:16.6),acs catalysis (2024, 14, 6981-6991;if:11.3),nano energy (2024, 126, 109698;if:16.8), nano energy (2024, 123, 109417;if:16.8), nano energy (2024, 121, 109249;if:16.8),applied catalysis b: environment and energy (2024, 357, 124269;if:20.2), applied catalysis b: environment and energy (2024, 351,123995;if:20.2)等国际期刊上。
首先,在理论计算预分析的指导下,王磊教授团队通过缺陷工程实现了nicop的电子再分布,使催化剂在碱性海水溶液中对析氢/析氧反应(her/oer)具有优异的催化活性和稳定性。pv降低了催化剂表面的电氧化重构能垒,使得在oer中更容易驱动晶体局部转化为活性羟基氧化物。采用nicopv@nf组装的阴离子交换膜(aem)电解槽具有高活性和长期稳定性,在100 ma cm-2时的工作效率高达77.0%,每加仑汽油当量的h2价格低至0.87美元,这为过渡金属磷化物在海水电解过程中的工业利用开辟了一条前景广阔的道路。(adv. energy mater. 2024, 2400975)
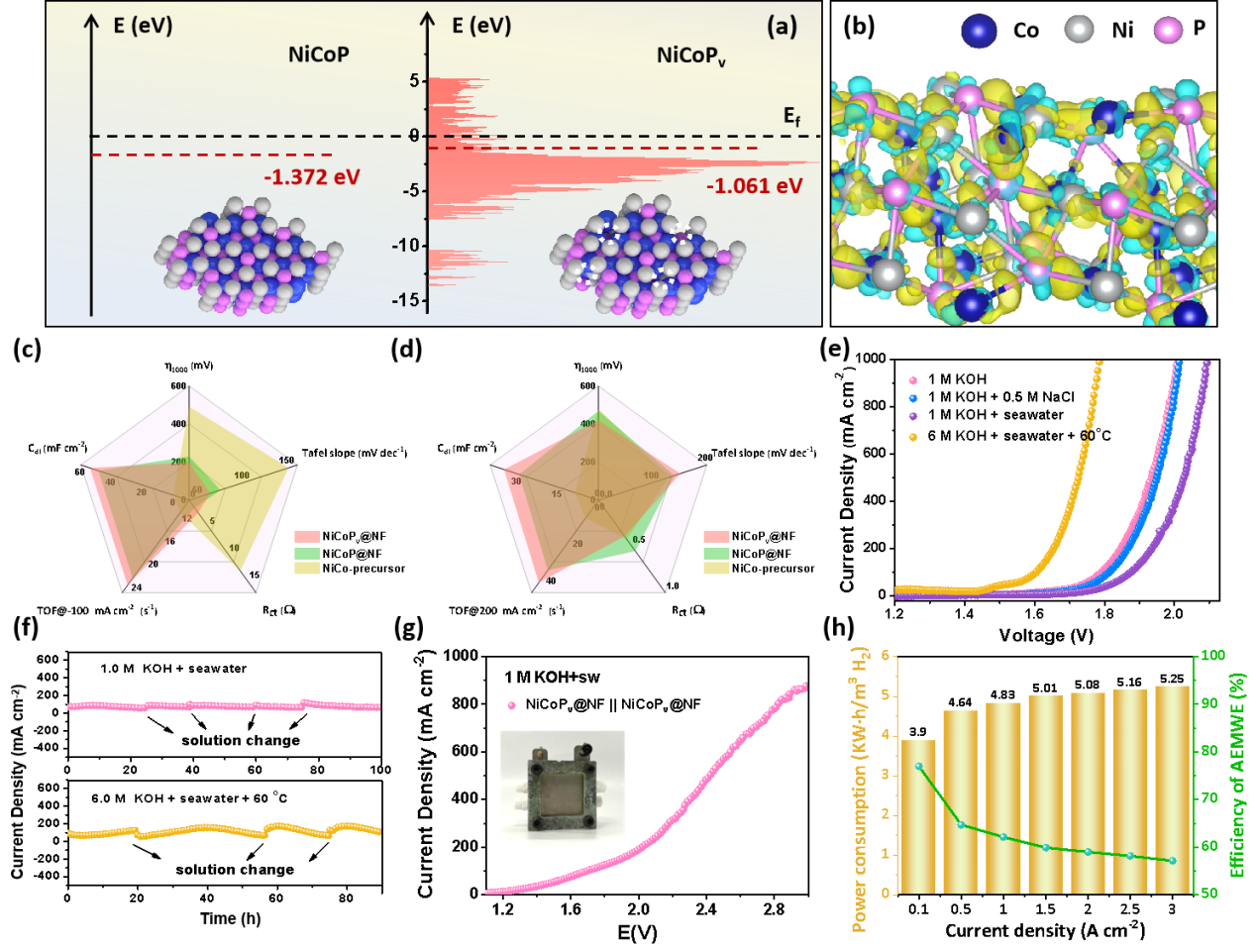
图1.nicopv@nf材料的性能及反应理论计算图。
成本低的fe3o4以其独特的八面体结构促进了水解离过程,但通常表现出较弱的氢中间体(hads)吸附能力,因此阻碍了her动力学。王磊教授团队通过ru/p双掺杂操纵了fe3o4的d带中心,弥补了fe3o4本身的电子缺陷。富电子的fe3o4表面表现出较高的hads覆盖率和趋于热中性的hads吉布斯自由能,这促使fe3o4突破了volmer步骤(决速步骤)的限制,成为了高效的海水析氢催化剂。此外,ru/p双掺杂显著降低了对cl-的吸附,提高了催化剂的防腐性能。(acs catal. 2024, 14, 6981-6991)
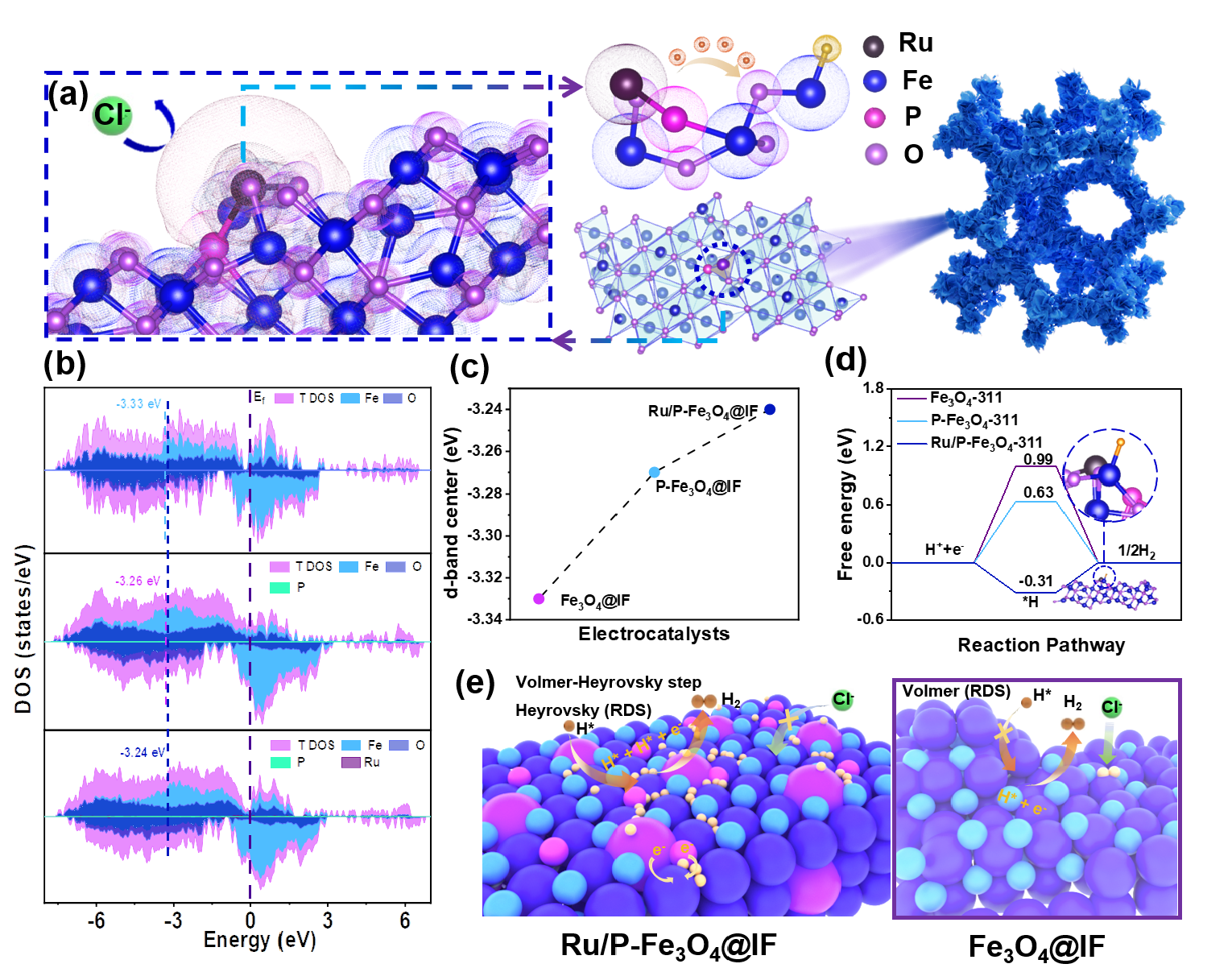
图2.ru/p-fe3o4@if材料的反应理论计算和her机制图。
此外,王磊教授团队采用离子掺杂和异质工程相结合的策略成功合成了在碱性海水溶液中对her具有高活性和高稳定性的p-pt/nimoo4@nf。实验证明,通过p原子掺杂增强了pt与nimoo4之间的强金属载体相互作用,优化了金属周围的环境,实现了高效、耐盐腐蚀的海水电解。理论计算表明,p原子掺杂可以调节电子结构,优化氢吸附自由能,降低cl-在催化剂表面的吸附。采用p-pt/nimoo4@nf作为阴极组装的aem电解槽在工业条件下仅以1.75 v即可驱动500 ma cm-2的电流密度,同时稳定运行上百小时。(nano energy, 2024, 126, 109698)
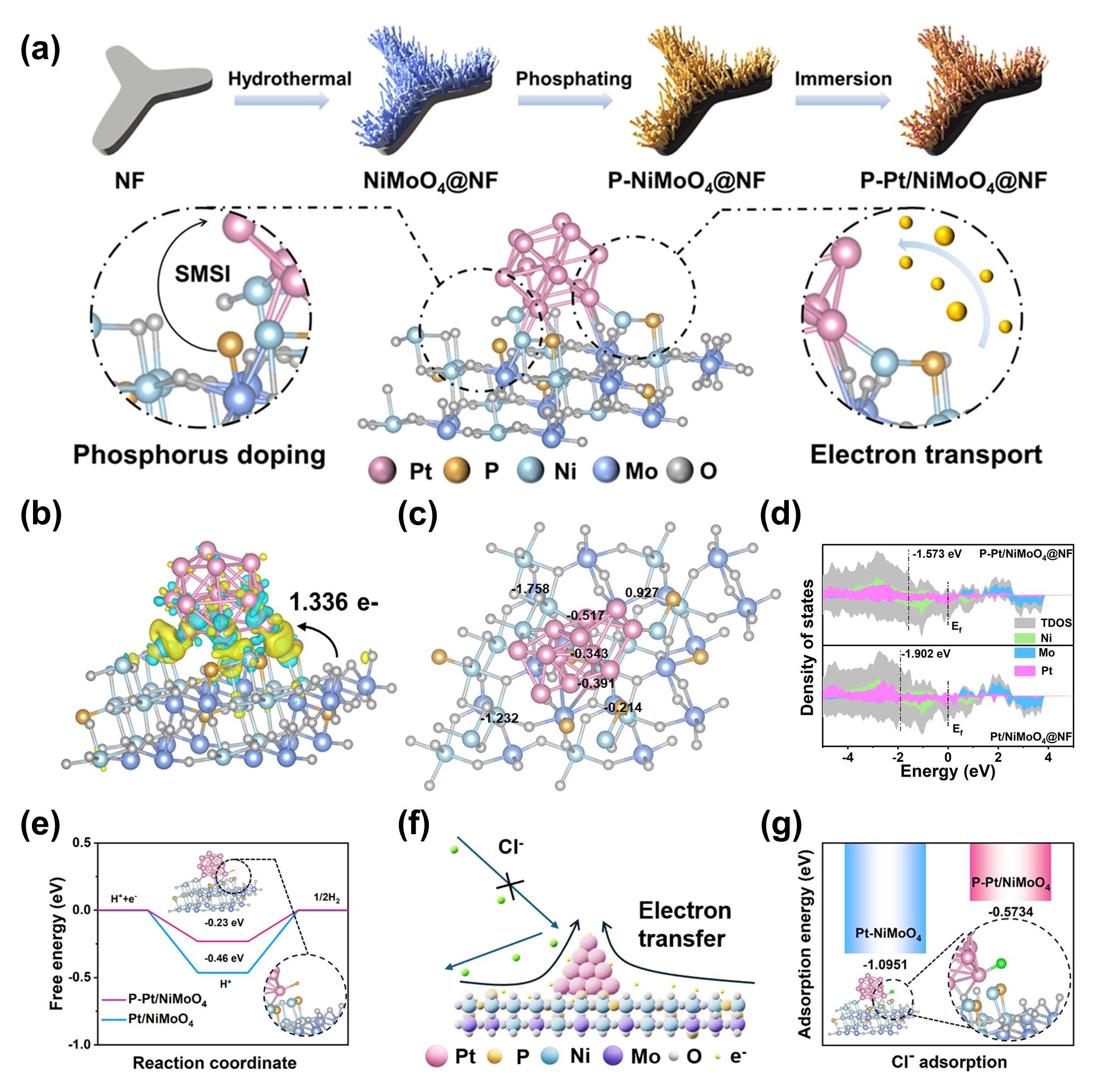
图3.p-pt/nimoo4@nf的强金属载体相互作用机理及反应理论计算图。
随后,王磊教授团队采用熔融盐与气相磷化相结合的策略构建了co/p双掺杂双功能催化剂(co/p-fe3o4@if),实现了fe3o4中电子的再分布。其中,磷化过程促进了fe2o3向反尖晶石fe3o4相的转化,加速了h2o的解离。同时,co掺杂操纵fe3o4中的电子分布,调控d带中心,在优化δgh*的同时促进了oer的重构过程。在碱性海水aem电解槽中,该催化剂在1.83 v电压下可以达到500 ma cm-2的电流密度,生产每立方米的氢气只需消耗4.38 kwh。(appl. catal. b: environ., 2024, 357, 124269)
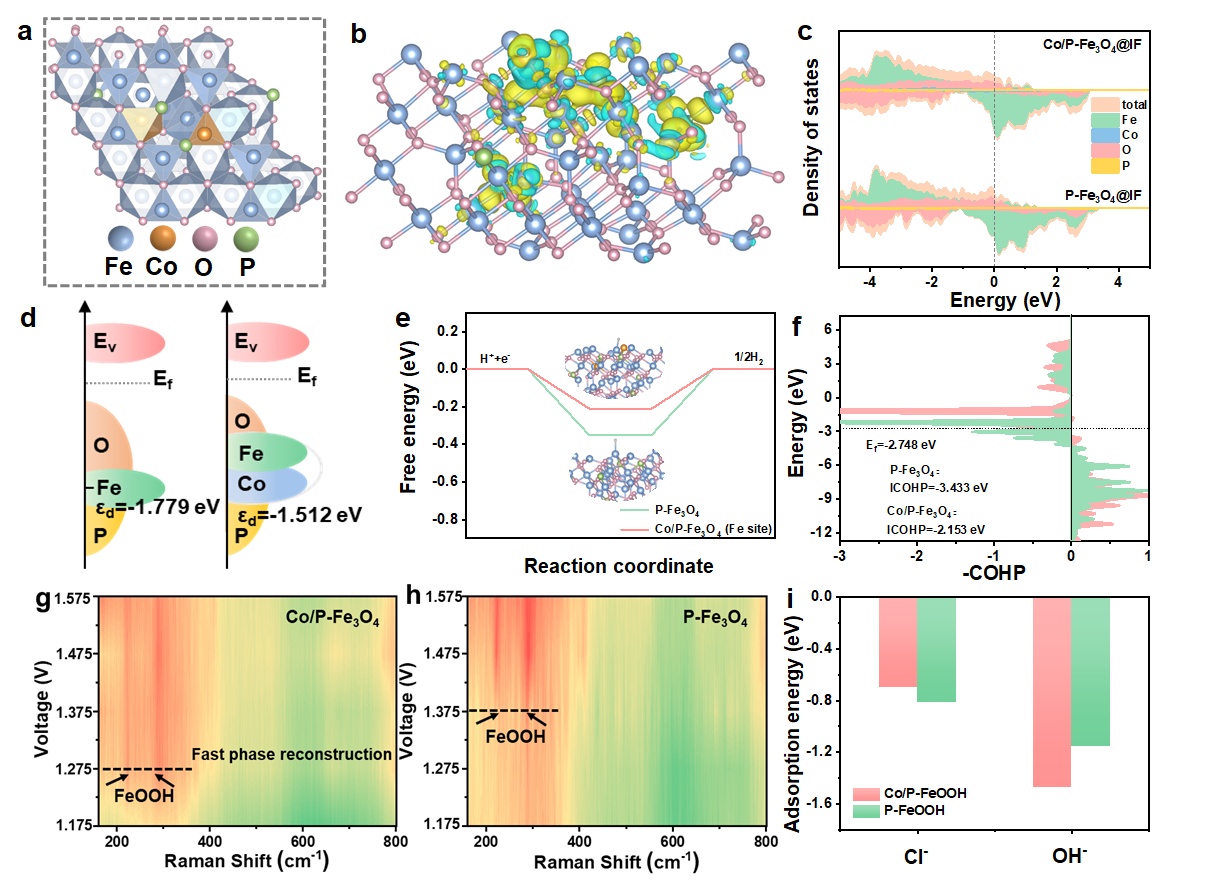
图4.co/p-fe3o4@if材料的反应理论计算图。
以上研究成果得到了国家自然科学基金、山东省青年泰山项目、山东省“外专双百计划”、山东省自然科学基金等项目的资助和支持。